Help & Examples for
Programs used by ENDscript are: SPDB to check residue numbering
and chainIDs from the input PDB1
file,
DSSP2
to calculate secondary structure elements, accessibility and disulphide
bridges, CNS3
to calculate intermolecular contacts,
BLAST4
to search for matching sequences in databases,
MULTALIN5
or CLUSTALW6
to perform multiple sequence
alignments, ESPript7
to gather all this information in PostScript figures,
BOBSCRIPT8 and
MOLSCRIPT9 to
display sequence conservation in 3D,
DINO to generate molecular surfaces colored according to sequence homology,
PROFIT to superimpose 3D structures of homolous sequences and
PHYLODENDRON to build a phylogenetic
tree.
These programs are executed in two phases.
To run the first phase you should connect to the interface, upload
your PDB file in the first box and click on SUBMIT in the
buttons frame. An ESPript figure is generated, giving information on each
monomeric sequence contained in your PDB file.
The second phase is launched by clicking on enable the BLAST search
in the second box of the interface and clicking again on SUBMIT.
A new ESPript figure is created, resulting
of the alignment of the sequence of the first monomer contained in your PDB file against
matching sequences. A third PostScript figure is generated with BOBSCRIPT, showing your 3D
structure coloured according to sequence similarity. Finally, the program check if an aligned
sequence has a known 3D structure, and each detected 3D structure is superimposed to the query
by the program PROFIT. A fourth PostScript is generated with BOBSCRIPT: the
query is rendered as a tube, its radius being proportional to the calculated rms deviation.
You can skip the first phase and run the second directly. ESPript and BOBSCRIPT
figures can be downloaded from the resulting frame of the interface.
Tracing files between programs
| First phase |
| |
programs |
input files |
output files |
| 1a |
SPDB |
file.pdb |
file.spdb |
| 1b |
DSSP |
file.spdb |
file.dssp |
| 1c |
CNS |
file.spdb |
file.ctct |
| 1d |
ESPript |
file.spdb file.ctct
file.dssp |
file1.ps |
| Second phase, part a |
| |
programs |
input files |
output files |
| 2a |
SPDB |
file.pdb |
file.seq |
| 2b |
BLAST and
MULTALIN or CLUSTALW |
file.seq |
file.aln |
1a
1b
1c |
SPDB
DSSP
CNS |
file.pdb
file.spdb
file.spdb |
file.spdb
file.dssp
file.ctct |
| 2c |
ESPript |
file.aln file.ctct file.spdb
file.dssp |
file1_bcol.pdb file1.bob
file2.ps |
| 2d |
BOBSCRIPT |
file1.bob file1_bcol.pdb |
file3.ps |
| Second phase, part b |
| |
programs |
input files |
output files |
| 2e |
PROFIT |
file.aln file.spdb
file_seq1.spdb file_seq2.spdb file_seq3.spdb... |
All rmsd |
| 2f |
BOBSCRIPT |
file2.bob file2_bcol.pdb |
file4.ps |
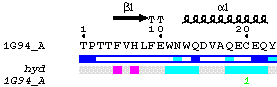
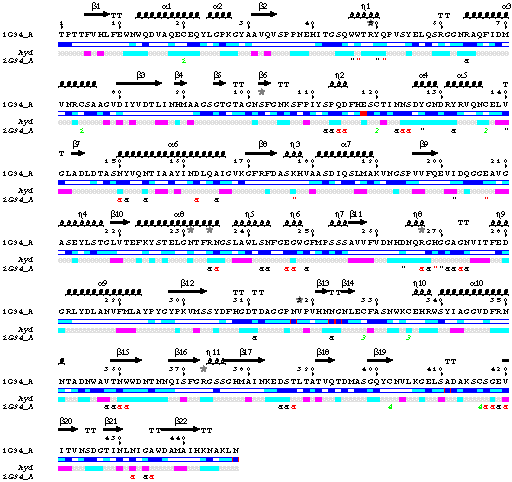
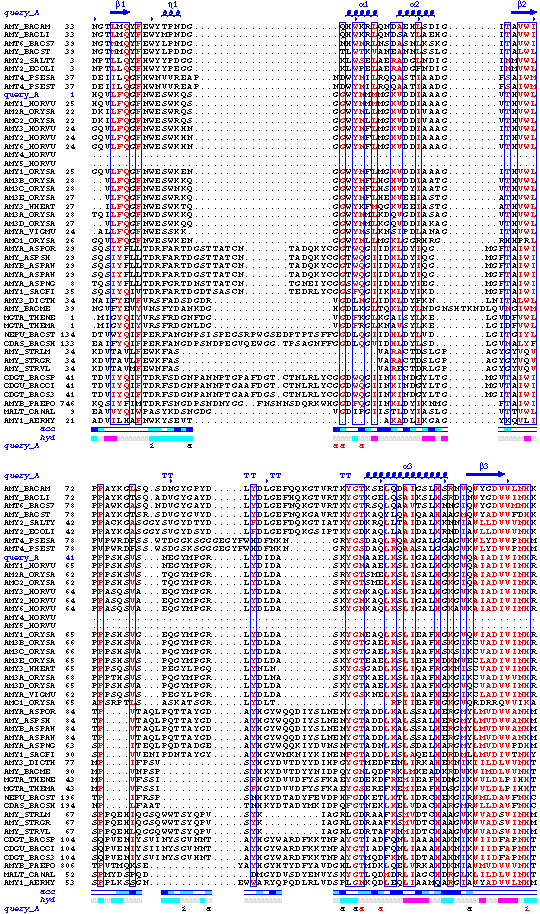
First phase: an ESPript figure showing all sequences of protein
contained in the input PDB file
1a. SPDB
| in -> out |
file.pdb -> file.spdb |
| main role |
checks input file |
SPDB (and by extension ENDscript) supports structure files downloaded
from the Protein Data Bank or resulting directly from the refinement programs
CNS and REFMACS10.
If necessary, the program renumbers protein residues and re-assign chainIDs
from
A to Z.
First model is kept for NMR structures.
First conformers are kept for alternate residues. They are flagged
by a * in column 68 of the output file.spdb.
Second oxygen atom of C-terminus main chain is removed (atom OXT).
Non-protein atoms are kept if they belong to:
-
nucleotide groups named ADE, GUA, CYT, THY or URI,
which are given the chainID
*
-
porphyrin groups named HEM, BCL, BPH or MQ7,
which are given the chainID
:
-
sugar groups named GLC, GAL, MAN, NAG, FUC, SIA or XYL,
which are given the chainID "
-
other ligands named NAD, NAH, NDP, NAP or FMN,
which are given the chainID
^
The user can keep other hetero-compounds contained in its PDB file.
He must type their names in the first box of the interface (up to 10
names of 2-3 characters per columns and one name per lines, check second example).
They will be renamed 0Ax, 0Bx, 0Cx or 0Dx by SPDB, where x is either a letter between A
and J or a digit between 0 and 9. An extra topology file needed by CNS
is generated in turn:
-
a group named 0A[A-J] is given the chainID *
a group named 0A[0-9] is given the chainID 1
-
a group named 0B[A-J] is given the chainID :
a group named 0B[0-9] is given the chainID 2
-
a group named 0C[A-J] is given the chainID "
a group named 0C[0-9] is given the chainID 3
-
a group named 0D[A-J] is given the chainID ^
a group named 0D[0-9] is given the chainID 4
Contacts between protein residues and kept hetero-compounds are shown
in turn in the PostScript figure, by using the chainIDs characters *,
%, !, ^, 1, 2, 3, 4, coloured
in red or black. By default, this mark corresponding to a protein-ligand contacts is shown
in red, if the distance is less than
3.2 Å, and in black if it is in the range 3.2-4 Å.
Remark: you can check the list of supported and unsupported
hetero-compounds included in your PDB file, by clicking on OUT
in the grey button panel when running the interface.
1b. DSSP
| in -> out |
file.spdb -> file.dssp |
| main role |
calculates secondary structure elements |
The program identifies alpha helices (shown by medium squiggles), 310 helices
(small squiggles), pi helices (large squiggles), beta strands (arrows), strict alpha turns
(TTT letters) and beta turns (TT letters) from the 3D structure.
Accessibility by residues is calculated. Only co-ordinates
of protein residues are taken into account.
Cysteins involved in disulphide bridges are extracted.
Residues with alternate conformations, flagged by a * in
column 68 of the input file.spdb, have this symbol
in column 138 of the output file.dssp.
If the option 'Display all known structures' is activated via the interface,
an automatic search is performed to check if a sequence name can be related to a known 3D structure.
This option has no effect in phase 1 but can be used in phase 2, when a BLAST
search is performed. Known secondary structure elements of each matching sequence are
displayed in turn in the ESPript figure.
1c. CNS
| in -> out |
file.spdb -> file.ctct |
| main role |
calculates intermolecular contacts |
CNS calculates both crystallographic and non-crystallographic contacts
between each protein molecule contained in file.spdb (remember that protein
residues are identified by A-Z chainIDs). Contacts between
protein residues and hetero-compounds are also calculated, if the latter
are identified by the chainIDs *, :, ", ^,
1, 2, 3 or 4.
Cell parameters and space group are extracted from the header
of file.spdb for crystallographic structures.
Hydrogen atoms are deleted and thus excluded from distance calculation.
Main chain atoms (N, CA, C, O) can
also be excluded from distance calculation, by clicking on a button in
the first box of the interface.
Upper limit for calculation of intermolecular contacts is 4 Å by default.
The shortest intermolecular distance is taken for each residue.
Command lines included in the script
to list intermolecular distances are:
Crystallographic contacts
Addition to CNS command file:
delete
selection=(hydrogen) end flags exclude * include pvdw end
parameter
nbond wmin=4.0 end end energy end
generates in CNS log file:
%atoms
"A -62 -ASN -OD1 " and "C -112 -THR -C "(XSYM# 4) only 3.64 A apart
Non-crystallographic contacts
Addition to CNS command file:
flags
exclude * include vdw end parameter nbond wmin=0 end end
for
$1 in (A B C D E F G H I J K L M N O P Q R S T U V W X Y Z) loop main
distance disposition=print cuton=0.0 cutoff=4
from =(segid $1) to =(not segid $1) end
end
loop main
generates in CNS log file:
atoms "A -90 -ALA -CB " and
"B -181 -HIS -CE1 " 3.6958 A apart
The line above shows a non-crystallographic vdw contact between carbon CB of Ala90 chainA
and carbon CE1 of His181 chainB.
Contacts can be further analysed by looking to the figure produced by
ESPript, as explained in the next paragraph.
1d. ESPript
| in -> out |
file.spdb file.ctct file.dssp
-> file1.ps |
| main role |
generates the first PostScript figure |
The protein sequence of each chainID contained in file.spdb is
displayed.
Alpha, 310 and pi helices are read from file.dssp.
They are shown above sequence in the PostScript as medium, small and large
squiggles with alpha, eta and pi labels respectively. Beta strands are shown
as arrows labelled beta. Strict alpha and beta turns are marked by TTT
and TT letters.
Residues modelled in file.pdb with an alternate conformation are
highlighted by a grey star on the top of sequences.
Relative accessibility is calculated from file.dssp. It is shown
by a blue-coloured bar below sequence. White is buried (<0.1) cyan is
intermediate (0.1-0.4) blue is accessible (>0.4) blue with red borders
is highly exposed (>1). A red box means that relative accessibility is not
calculated for the residue, because it is truncated.
Hydropathy is calculated from the sequence according to the algorithm of
Kyte and Doolittle11. It is shown by a
second bar below accessibility: pink is hydrophobic, grey is intermediate
and cyan is hydrophilic.
Disulphide bridges are extracted from file.dssp. They are shown
by green digits (1 1, 2
2 ...) below the bar of hydropathy.
Intermolecular contacts are extracted from file.ctct and are
displayed along with disulphide bridges below the bar of hydropathy. The shortest intermolecular distance
is taken for each residue. Corresponding chainIDs are written in
red
if the distance is less than 3.2 Å and in black if the distance
is in the range 3.2-4 Å.
Further information is given according to the font:
-
a to z in italic are
crystallographic contacts between residues
-
A to Z in italic are
non-crystallographic contacts between residues
-
# identifies a crystallographic contact between two
residues having same names, numbers and chainIDs (crystallographic identity)
-
a to z are crystallographic contacts
between residues having same names and numbers but different chainIDs.
-
A to Z are non-crystallographic contacts
between residues having same names, numbers and different chainIDs (for
example along a non-crystallographic 2-fold axis).
-
*, :, ", ^, 1, 2, 3, 4
are contacts between protein residues
and supported hetero-compounds.
Thus, if in file.ctct the line:
atoms "A -90 -ALA -CB " and
"B -181 -HIS -CE1 " 3.6958 A apart
corresponds to the shortest distance between Ala90 and His181. A black
B is written below Ala90 in the sequence of chainA.
Remark: only molecules located in the crystallographic asymmetric unit
are taken into account by DSSP in its calculation of accessibility.
Thus, you can find 'highly accessible' residues involved in contacts with
crystallographic neighbours according to the ESPript figure. These residues
are in fact buried in the crystal lattice.
Output Layout
-
FontSize [default: 6]
-
Size in points for the fonts (Courier for sequence
names and residues).
-
ColumnNb [default: 90]
-
Number of residue columns per line.
- A value for column number is calculated in the
log file, at the end of the section concerning ESPript (click on OUT in the grey buttons panel
and look for the sentence 'suggestion columns per line').
Replace the default, 90, by this value and click on SUBMIT so as to obtain a
justified figure.
-
Vgap [default: 7]
-
Vertical gap between two blocks of sequences. The
unit for the distance is the height of a line.
-
Vshift [default: 0]
-
Vertical shift for the whole display. The unit for
the distance is the height of a line.
-
Hshift [default: 0, centred]
-
Horizontal shift for the whole display. The unit
for the distance is the width of a residue.
-
PrinterOpt [default: C]
-
C coloured output ; T coloured with all
letters in bold, ideal for thermal printers and others before reduction of your figure in an article ; S light yellow
background, ideal for slides ; B black & white, a grey scale is used ; F flashy
colours, similar residues are written with black bold characters and boxed in yellow, ideal for overheads.
-
Paper [default: P]
-
P: Portrait(A4) ; L: Landscape(A4)
; P3: Portrait(A3) ; L3: Landscape(A3).
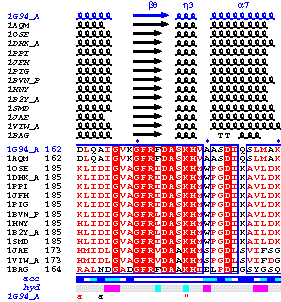
Second phase, part a: a multiple alignment colour-coded with ESPript
and a 3D representation obtained with BOBSCRIPT
2a. SPDB
| in -> out |
file.pdb -> file.seq |
| main role |
checks input file |
SPDB (and by extension ENDscript) supports structure files downloaded
from the Protein Data Bank or resulting directly from the refinement programs
CNS and REFMACS.
The sequence of the first molecule is extracted and written in a one character
code in FASTA format.
2b. BLAST and MULTALIN or CLUSTALW
| in -> out |
file.seq -> (file.blast) ->
file.aln |
| main role |
finds homologous sequences for multiple
alignment |
The BLAST search can be performed against sequences extracted from the PDB (named pdbaa) or against
the SWISSPROT or the TrEMBL12 databases. It can also
be performed against SEQUENCED GENOMES downloaded from the
ExPASy server.
The comparison matrix is BLOSUM6213 and
the threshold for the evalue is set to 10-6.
Multiple alignments are performed by MULTALIN or CLUSTALW.
A FAST method is used with CLUSTALW.
Output sequences are in the same order as they have been aligned from the guide tree.
They can also be in the same order as they have been entered, from the lowest
to the highest evalue, if the option input is used in the interface.
You can edit outputs from BLAST with a short description of each sequence by clicking in the
Results frame of the interface, section Tracing files.
If activated, the option 'Show strictly conserved side chains' allows to display side chains
of strictly conserved residues with a blue colour in the BOBSCRIPT figure.
If needed, BLAST and CLUSTALW searches can be cross-checked using the
NPS@14 server,
to have a better control on defaults or to use other sequence databases. Resulting alignment files
in MPSA format can be uploaded in ESPript.
2c. ESPript
| in -> out |
file.aln file.spdb file.ctct file.dssp
-> file1_bcol.pdb file1.bob file2.ps |
| main role |
generates a second figure in PostScript
with a multiple alignment |
Similarities between the PDB sequence of the chosen chainID (chainA by
default) and homologous sequences aligned with CLUSTALW
are rendered by a boxing in colour. A score is calculated for each column of residues, according
to a matrix based on physico-chemical properties. Residue names are written in black
if score is below 0.7 (low similarity); they are in red and
framed in blue if score is
in the range 0.7-1 (high similarity); they are in white on a red background
in case of strict identity.
You can switch to other scoring matrices using the html form and re-run
phase 2. A Risler matrix15 gives usually an excellent
rendering, when showing similarities on the 3D structure using BOBSCRIPT.
- Risler, Blosum62,
Pam250
and Identity are four possibilities of scoring matrix (check appendix in the ESPript manual).
- A percentage of Equivalent residues
can also be calculated considering either physico-chemical properties
(HKR are polar positive ; DE are polar negative ; STNQ
are polar neutral ; AVLIM are non polar aliphatic ; FYW
are non polar aromatic ; PG ; C) or similarities used in
MULTALIN
(IV ; LM ; FY ; NDQEBZ).
Secondary structure elements, relative accessibility, hydropathy and intermolecular
contacts are displayed as in file1.ps.
Sequences can be removed or their order can be changed by using the box
'Defining group' (e.g. 1-15 18-47 removes sequences
16 and 17 as shown in the second example).
Similarity scores by residues are written in the bfactor column of an output
file named file1_bcol.pdb. This PDB file includes the co-ordinates
of the selected protein chain and of its bound hetero-compounds, according
to the list of interactions read in file.ctct. In the meantime, the command
file file1.bob is created for BOBSCRIPT. file1.bob
contains a script for representation of secondary structure elements
and reads file1_bcol.pdb.
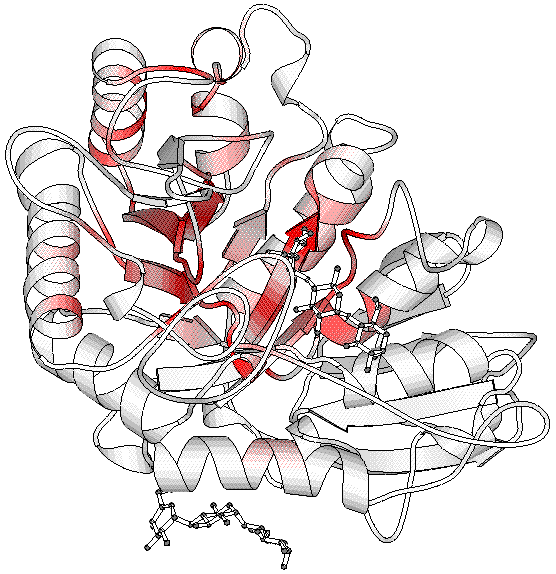
2d. Similarities on 3D structure with BOBSCRIPT
| in -> out |
file1.bob file1_bcol.pdb -> file3.ps |
| main role |
generates a PostScript figure of the 3D structure |
Secondary structure elements previously determined by DSSP
(helices and beta strands) are shown. They are colour-coded from white to red
according to similarity scores (low to high).
By default, side chains of strictly conserved residues are shown in blue,
if the button 'Show strictly conserved side chains' has been activated in the
section 'Start a BLAST request' of the interface.
A green dashed line links CA atoms of two cysteins
connected by a disulphide bridge.
Hetero-compounds in interaction with the selected protein chain are represented
by grey ball-and-sticks. Note that only hetero-compounds located in the
asymmetric unit are displayed for crystallographic structures.
Second phase, part b: a 3D representation with rmsd information
2e. PROFIT
| in -> out |
file.aln file.spdb file_seq1.spdb file_seq2.spdb file_seq3.spdb... ->
All rmsd |
| main role |
superimpose homologous structures to the query |
Information on zones of equivalent residues is extracted by PROFIT from file.aln,
so as to superimpose each known structure of aligned sequence onto the query. Thus, each mobile
structure (file_seq1.spdb, file_seq2.spdb...) is fitted onto the reference structure
(file.spdb) by using CA pairs. Fitted structures are written in turn in a tar file named All.
A rmsd by residue is calculated using all fitted CA pairs
2f. RMSD on 3D structure with BOBSCRIPT
| in -> out |
file2.bob file2_bcol.pdb -> file4.ps |
| main role |
generates a PostScript figure with rmsd information |
A new file named file2_bcol.pdb
is created, which contains a rmsd score by residue in the occupancy column
and a similarity score in the temperature factor column. This information is
used in the BOBSCRIPT command file file2.bob, in order to generate a
ribbon representation of the query: the protein is represented as a tube,
its radius being proportional to the calculated rmsd. A colour ramping from white
to red is still used to visualize variations in sequence similarity (from low to high).
Critical command line in file2.bob allowing such representation is:
colour ss from rgb .2 .2 .2 via white to red by b-factor from -100 to 100;
set coilradius from .2 to 1.2 by occupancy from .2 to 5;
Appendix
Three supplementary links in phase 2
1. MOLSCRIPT
| in -> out |
file1.bob file1_bcol.pdb ->
file3.vrml |
| main role |
rotates a 3D structure with colour-coded
similarities |
A MOLSCRIPT figure in VRML is also created in phase 2. It can be displayed and
rotated on your screen, if your web browser has
the appropriate plug-in from CORTONA
or COSMO.
For information, command files are similar to generate MOLSCRIPT and BOBSCRIPT figures.
However the line in file1.bob defining the colour code is specific to each
program.
!!COLOUR IN MOLSCRIPT!!
!set colourparts on, residuecolour amino-acids b-factor 0 100 from blue to red;
!!COLOUR IN BOBSCRIPT!!
colour ss from white to red by b-factor from 0 to 100;
The above line is uncommented in MOLSCRIPT and the VRML file is generated using the
command
molscript -vrml < file1.bob > file3.vrml
2. PHYLODENDRON
A click on the icon with a tree in the Results frame allows to generate and view phylogenetic trees, if you have used
CLUSTALW as alignment program. Just press the button 'Submit', once PHYLODENDRON's interface
appears on your screen.
3. ESPript
You can have access to the full ESPript
interface by clicking on the ESPript button of the interface.
This form allows you to have a better grip on the layout, to highlight important zones and residues
and so on...
Thus, you can find in the box 'Special Characters' of the interface
the script.
X B query_A ! secondary structure elements are coloured in blue
Y B query_A ! residue numbering is in accord with the sequence query_A
and is coloured in blue
P B query_A ! hydropathy is calculated, 'hyd' is in blue
T B query_A ! sequence name is in blue
You can also obtain a figure with full sequences, by replacing the
range typed in the first box of the interface (eg 4-4500) by the string all
Examples
1. One monomer in the asymmetric unit and one supported hetero-compound
The first example refers to the structure of a psychrophilic alpha amylase from
bacteria, solved by Nushin Aghajari16 in the group
of Richard Haser, Laboratoire
de BioCristallographie, IBCP Lyon. It is deposited with the PDB under
the code 1G94. The protein is made of 448 residues and catalyses the hydrolysis of polysaccharides.
It crystallizes in space group C2221 with one molecule in the asymmetric unit. Two
bound glucoses named GLC are included in the PDB file, in addition to other sugar groups
which are unsupported by ENDscript by default.
To start with the interface, either type the code 1G94 in the
first box named 'pdb data file' or click on the PDB icon of this box, type 1G94 and click on 'retrieve file'.
Click on SUBMIT in the bottom left command panel to generate the first PostScrit output.
- The sequence is displayed along with secondary structure elements, hydropathy and
crystallographic contacts.
- Seven alternate conformations have been detected and are marked by grey stars above sequence.
- Accessibility of Glu118 is in red, because this residue is disordered and the co-ordinates
of its side chain beyond CB atom are not entered in the PDB file.
- Contacts between the protein and the two GLC molecules are marked by a "
on the same line, as well as crystallographic contacts.
Four disulphide bridges are shown with green digits.
Remark: Note, that the list of contacts can be checked by: (i) clicking on
the link 'CNS' in the section Tracing text files of the Results frame;
(ii) searching in the CNS file the string %atoms at the
beginning of each line of non-crystalographic contacts
and the string atoms at the beginning of each line of
non-crystalographic contacts.
Change the threshold from 10-6 to
10-12, switch from MULTALIN to CLUSTALW,
click on BLAST to start the search and to extract high homologous sequences from the PDB.
- Fourteen similar sequences have been detected, including 1G94 itself. According to sequence similarity,
one can notice that the alignment is certainly false at the second disulphide bridge of 1G94
(2 2) and should be manually corrected.
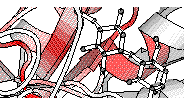
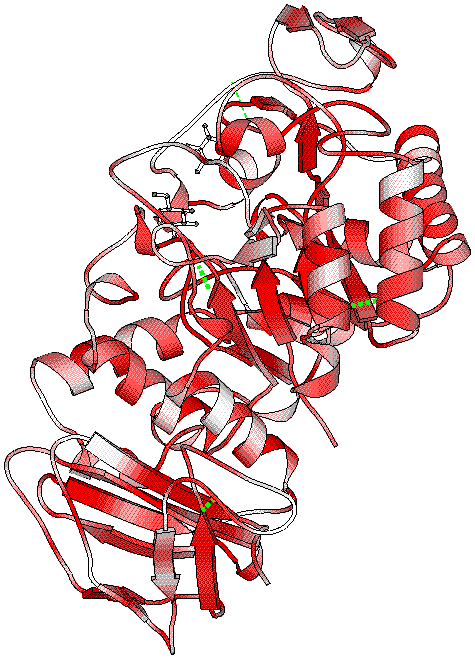
The third PostScript figure has been generated, showing similarities on the structure file.
- CA atoms of the four disulphide bonds are connected by green dashes
and two glucose molecules are visible at the surface of the protein.
- The command file for BOBSCRIPT is similar to the one for
MOLSCRIPT. It can
be copy-paste after a click on the IN of the buttons frame, but the line defining the colour code must be changed.
!!COLOUR IN MOLSCRIPT!!
set colourparts on, residuecolour amino-acids b-factor 0 100 from blue to red;
!!COLOUR IN BOBSCRIPT!!
!colour ss from white to red by b-factor from 0 to 100;
You can rotate the molecule using your web browser by clicking on the VRML file
generated by MOLSCRIPT in the RESULTS FRAME,
or you can retrieve the PDB file created by ESPript with similarity scores on the bfactor column
(hyperlink BCOL in the Results frame) to display the protein on a Silicon Graphics
workstation.
molscript -gl < file1.bob
The protein can also be rotated and a new transformation matrix can be chosen.
Finally, homologous structures are superimposed to the query by the program PROFIT and
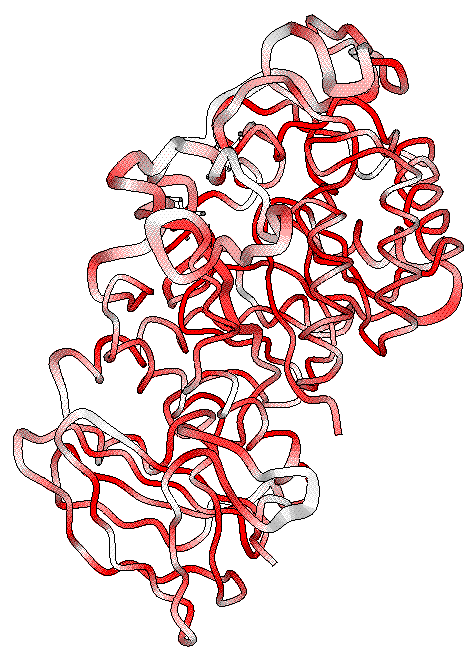
the fourth PostScript figure is generated, showing rmsd information.
- As can be expected, zones at the protein surface exhibit higher rmsd values (larger radius)
and lower similarity scores (white colour).
2. Two monomers in the asymmetric unit and keeping unsupported hetero-compounds
The second example refers to the structure of alpha-amylase from barley,
isoenzyme 1, solved by Xavier Robert
in the same laboratory. This alpha amylase, known as amy1, can crystallize
in space group P212121
with two molecules in the asymmetric unit. Each monomer binds
molecules of acarbose, an inhibitor for the
hydrolysis reaction. Input file is query.pdb. It contains
2x405 residues (protein is truncated and the first 24 residues are
missing), 2x2 molecules of inhibitor
named ACR and ACE, calcium ions named CA and water molecules named HOH.
These hetero-compounds are not supported by ENDscript by default.
Save query.pdb on your disk,
click on the button EXIT then EXECUTE of the html
interface
if you are still dealing with the first example.
Click on 'Browse' in the first box to upload the PDB file.
We want to keep acarbose during analysis by ENDscript.
The names ACR, ACE are typed in the columns with symbols 1, 2
contained in the box 'Keeping hetero-compounds'.
In the box 'Start a BLAST request': switch from the default database PDBAA to the SWISSPROT, from MULTALIN to CLUSTALW, release button Display
all known structures (just for convenience for the example), click on button enable
the BLAST search and click on SUBMIT. The three
PostScript figures will be obtained in one go after a few minutes of calculation.
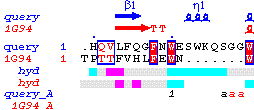
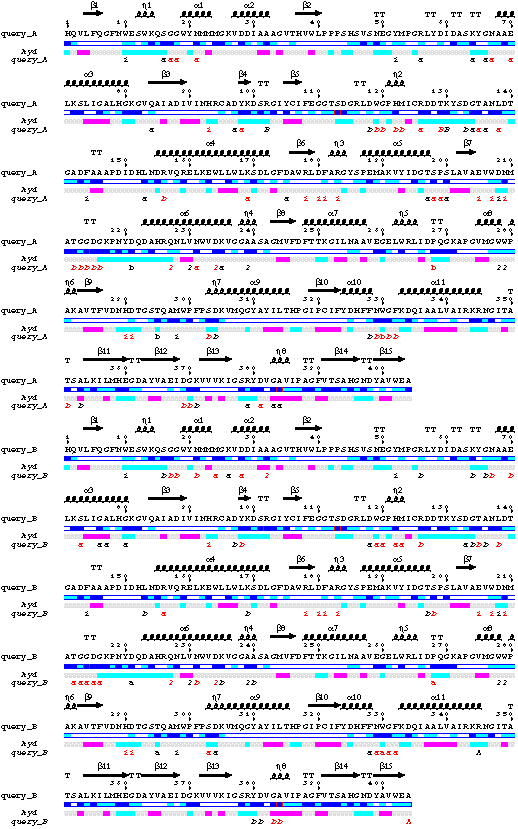
- The sequences of both chains A and B are displayed on the figure.
-The line query_A corresponds to the contacts made by molecule A: i.e.
crystallographic contacts with residues of molecules A and B (a,
b), molecular contacts with molecule B (B), crystallographic
contacts of Pro268 and Trp330 against their own symmetric (b), both
crystallographic and non-crystallographic contacts with acarbose molecules (1 2).
- Sequence of chainA has been extracted. It appears in position 9 after
the multiple alignment performed by CLUSTALW. Sequence 10 named AMY1_HORVU corresponds
to the sequence of amy1 deposited with the SWISSPROT. Therefore, it starts at residue 25.
Its is clear from the PostScript figure, that sequences 16 and 17 are fragments.
Type in the box 'Defining group' of the interface:
1-15 18-47
and click on SUBMIT in the button panel. The three figures are
re-created without sequences 16 and 17.
Two residues are now strictly conserved in all sequences. One of them, Asp180,
is an essential catalytic residue and binds acarbose molecule 1.
The third figure was obtained with BOBSCRIPT.
The image shows that molecule 1 is bound in a well conserved region, i.e. the
active site of the protein, while molecule 2 is bound away
in a non-conserved region. However, the multiple alignments figure suggests that this peripheral binding site is conserved
in plant alpha amylases.
Finally, we want to generate an ESPript figure with intermolecular contacts made by
monomers A and B.
-click on the ESPript button of the interface for connection. The script concerning
the second ENDscript figure is transferred.
-add in the box 'Special characters' of ESPript, under T B query_A
R A all
R B all
Note that this request is relevant
only if A and B have the same sequence. Click on the SUBMIT button:
-Contacts with acarbose are conserved in the two monomers.
3. Superposing mesophilic and psychrophilic alpha-amylases
This section concern users having already well practiced ESPript. DSSP and CNS output files
have been saved on the disk and we want to compare the two sequences of
alpha-amylases. Similarities are poor between the two proteins, 15% of identity
in sequence, but they share the same fold and
13 matching segments of 3-33 residues were detected after structural superposition
by the program TOP17.
The two sequences were aligned manually using both
this information and the editor
SEAVIEW18.
The following figure was obtained in turn with ESPript:
The main frame has been duplicated by clicking on +1 in the
button panel; files concerning barley alpha-amylase have been entered in the upper part
[01]
and files concerning 1G94 in the bottom part [01].
A vertical shit of -1 in [01]
and a bottom shift of -1 in both [01]
and [01] allow to display
all information on secondary structure elements, hydropathy and intermolecular contacts
in the resulting PostScript. The essential Asp180 is highlighted by a blue box. 81 columns have been used instead of 70,
in order to obtain a justified figure as suggested at the end of the OUT file.
Structural similarities are obvious on the figure despite the lack of sequence conservation, but there
is no striking feature explaining the psychrophilic specificity of 1G94.
References
1. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat,
T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. (2000)
The Protein Data Bank. Nucleic Acids Res. 28 235-242
2. Kabsch, W. and Sander, C. (1983)
Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded
and geometrical features. Biopolymers. 22 2577-2637
3. Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L.,
Gros P., Grosse-Kunstleve, R.W., Jiang J.S., Kuszewski, J., Nilges, M.,
Pannu, N.S., Read, R.J., Rice L.M., Simonson, T. and Warren, G.L. (2000)
Crystallography & NMR system: A new software suite for macromolecular
structure determination. Acta Crystallogr. D. 54 905-921
4.Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang J., Zhang,
Z., Miller, W. and Lipman, D.J. (1997)
Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs. Nucleic Acids Res. 25 3389-3402
5. Corpet, F. (1988)
Multiple sequence alignment with hierarchical clustering.
Nucl. Acids Res. 16 10881-10890
6. Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994)
CLUSTAL W: improving the sensitivity of progressive multiple sequence
alignment through sequence weighting, positions-specific gap penalties
and weight matrix choice. Nucl. Acids Res. 22 4673-4680
7. Gouet, P., Courcelle, E., Stuart, D.I. and Metoz, F. (1999)
ESPript: multiple sequence alignments in PostScript. Bioinformatics.
15 305-308
8. Esnouf, R.M. (1997)
An extensively modified version of MolScript that includes greatly
enhanced coloring capabilities. J. Mol. Graphics. 15 132-134
9. Kraulis, P.J. (1991)
MOLSCRIPT: a program to produce both detailed and schematic plots
of protein structures. J. Appl. Cryst. 24 946-950
10. Murshudov, G.N., Vagin. A.A. and Dodson, E.J. (1997). Refinement
of macromolecular structures by the maximum-likelyhood method
Acta Crystallogr. D. 53 240-255
11. Kyte, J. and Doolittle, R. (1982)
A simple method for displaying the hydropathic character of a protein.
J. Mol. Biol. 157 105-132
12. Bairoch, A. and Apweiler, R. (1997)
The SWISS-PROT protein sequence data bank and its supplement TrEMBL.
Nucleic Acids Res. 25 31-36
13. Henikoff, J.G. and Henikoff, S. (1996)
Blocks database and applications. Meth. in Enzym. 266
88-105
14. Combet, C., Blanchet, C., Geourjon, C. and Deleage, G. (2000)
NPS@: Network Protein Sequence Analysiss.
TIBS. 25 147-150
15. Risler, J.L., Delorme, M.O., Delacroix, H. and Henaut, A.
(1988)
Amino acid substitutions in structurally related proteins. A pattern
recognition approach. Determination of a new and efficient scoring matrix.
J.
Mol. Biol. 204 1019-1029
16. Aghajari, N., Roth, M. and Haser, R. (2001)
Enzymatic synthesis in the crystalline state of a novel hepta-saccharide:
its 3D-structure in complex with a cold active alpha-amylase
submitted
17. Lu, G. (2000)
TOP: a new method for protein structure comparisons and similarity searches.
J. Appl. Cryst. 33 176-183
18. Galtier, N., Gouy, M. and Gauthier, C. (1996)
SeaView and Phylo_win, two graphic tools for sequence alignment and
molecular phylogeny. Comput. Applic. Biosci. 12 543-548
Back to MAIN page.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}