BACKGROUND
Variability
Sequence variability within related protein sequences has
long ago been recognized to show significant clues about their 3-dimensional
structure and function. Wu and Kabat (1977) were the first to define a
variability coefficient that led them to predict that
segments of high amino acid variability in immunoglobulins
correspond to their antigen binding
sites. Thus, high variability regions in a group of related proteins are linked to the
specificity of the molecules. On the other hand, regions with low variability are usually structurals,
or define regions of common function.
Shannon Entropy
Shannon entropy analysis (Shannon, 1942) is possibly the most sensitive
tool to estimate the diversity
of a system.
For a multiple protein sequence alignment
the Shannon entropy (H) for every position is as follow:
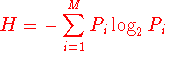
Where Pi is the fraction of residues of amino acid type
i, and M is the number of amino acid types (20).
H ranges from 0 (only one residue in present at that position) to 4.322
(all 20 residues are equally represented in that position). Typically,
positions with H >2.0 are considerered variable, whereas those with H < 2 are consider conserved. Highly conserved positions are those with H <1.0 (Litwin and Jores, 1992).
A minimum number of sequences is however required (~100) for H to describe the
diversity of a protein family.
Motivation
Sequence variability through mutation and subsequent immune
selection (a process called "antigen drift") is also a common mechanism by which
some viruses (AIDS, Influenza) and other pathogens (Plasmodium, Trypanosomatides, etc)
escape the immune system. Thus, for vaccine design, it is imperative to focus on protein fragments of
low variability (Shannon Entropy). With this in mind, we have implemented a
"Shannon Filter" functionality on the server that identifies
sequence stretches from the protein alignment whose values of variablity (H) are
under a given value, which is set by the user.
USERGUIDE
Input
The input for this server must be a multiple sequence alignment in Clustal
format. Only the standard 20 amino acids should be included in the
alignment. If other sequence characters are included (e.g. X) the server
will return an error message.
A typical example of Clustal alignment is the following:
CLUSTAL W (1.81) multiple sequence alignment
hla_a68w_1HSB SHSMRYFYTSVSRPGRGEPRFIAVGYVDDTQFVRFDSDAASQRMEPRAPWI
hla_a0201_1DUY SHSMRYFFTSVSRPGRGEPRFIAVGYVDDTQFVRFDSDAASQRMEPRAPWI
hla_b3501_1A1N SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRTEPRPPWI
hla_b5301_1A1M SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRTEPRPPWI
hla_b5101_1E27 SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRTEPRAPWI
hla_b2701_1HSA SHSMRYFHTSVSRPGRGEPRFITVGYVDDTLFVRFDSDAASPREEPRAPWI
hla_cw3_1EFX SHSMRYFYTAVSRPGRGEPHFIAVGYVDDTQFVRFDSDAASPRGEPRAPWV
hla-cw4_1IM9 SHSMRYFSTSVSWPGRGEPRFIAVGYVDDTQFVRFDSDAASPRGEPREPWV
mkb_2vaa PHSLRYFVTAVSRPGLGEPRYMEVGYVDDTEFVRFDSDAENPRYEPRARWM
db-1BZ9 PHSMRYFETAVSRPGLEEPRYISVGYVDNKEFVRFDSDAENPRYEPRAPWM
.**:*** *::* ** **::: *****:. ******** . * *** *:
hla_a68w_1HSB RNTRNVKAQSQTDRVDLGTLRGYYNQSEAGSHTIQMMYGCDVGS
hla_a0201_1DUY GETRKVKAHSQTHRVDLGTLRGYYNQSEAGSHTVQRMYGCDVGS
hla_b3501_1A1N RNTQIFKTNTQTYRESLRNLRGYYNQSEAGSHIIQRMYGCDLGP
hla_b5301_1A1M RNTQIFKTNTQTYRENLRIALRYYNQSEAGSHIIQRMYGCDLGP
hla_b5101_1E27 RNTQIFKTNTQTYRENLRIALRYYNQSEAGSHTWQTMYGCDVGP
hla_b2701_1HSA RETQICKAKAQTDREDLRTLLRYYNQSEAGSHTLQNMYGCDVGP
hla_cw3_1EFX RETQKYKRQAQTDRVSLRNLRGYYNQSEAGSHIIQRMYGCDVGP
hla-cw4_1IM9 RETQKYKRQAQADRVNLRKLRGYYNQSEDGSHTLQRMFGCDLGP
mkb_2vaa RETQKAKGNEQSFRVDLRTLLGYYNQSKGGSHTIQVISGCEVGS
db-1BZ9 RETQKAKGQEQWFRVSLRNLLGYYNQSAGGSHTLQQMSGCDLGS
:*: * : * * .* ***** *** * : **::*.
Options
Sequence SettingsIn addition to the sequence variability analysis, the server also derives
a consensus sequence from the input multiple sequence alignment
at threshold choices (50, 70, 80 and 90%) set by the users. Upper capital
letters in the consensus sequence represent the standard amino acids, and
lower capital letter are for the following amino acid groups:
o alcohol = S, T
- negative = D, E
s small = G, A, S
+ positive = H, K, R
a aromatic = F, Y, W
x hydrox = alcohol + Y
l aliphatic = I, V, L, A
c charged = positive & negative
h hydrophobic = aromatic, aliphatic & [A, M, C]
p polar = charged, alcohol & [Q, N, C]
. all = G, A, V, I, L, M, F, Y, W, H, C, P, K, R, D, E, Q, N, S, T
For the output the user can set any of the following sequences as the reference:
- The consensus sequence
- The most abundant sequence, resulting of taking the most frequent
residue at every position in the alignment.
- The first sequence in the sequence alignment. This is especially useful if
the user has additional information on a given sequence, and wants to set it as the standard
Shannon FilterThe Shannon Filter identifies sequence stretches
with a variability under a given value of H. The "Shannon Filter" takes
two parameters: Fragment size and Shannon Entropy Threshold.
- Shannon Entropy Threshold:
This parameter has to be set within the
range of the Shannon Entropy values (0 to 4.3), and only one decimal is allowed. Those positions with a value of H above the threshold
are filtered out. The default value is set to 1.3. Positions with H <
1.3 are considered of low variability (highly conserved).
- Fragment Length:
This parameter set the minimun length of the fragment. Each of the fragment
residues has a H that is under the threshold value
Only the longest
stretch of residues with H under the threshold is listed.
Output
The output generated by the server consists of three parts:
1) A graph of the Shannon Entropy plotted against
the selected choice of sequence: consensus, most abundant, or first sequence.
For a better display of the graphics, the input alignment should not exceed 200
letters.
2) The results of the Shannon Filter showing the sequence stretches that
had a minimun number of consecutive residues - set by the the user- with Shannon Entropy
values under or equal the threshold. The fragments are sorted in a table where
the initial and final positions of the fragment in the alignment are shown,
as well as their amino acid sequence.
An example:
| Fragments of 8 or more consecutive residues with H < 1.3 |
| N | Start | End | Sequence |
|---|
| 1 | 12 |
20 | N E K D L L A L D |
| 2 | 30 |
38 | I T N W L W Y I K |
3) The data stored in the server for only 5 days, and it can
be retreived from the relevant link.
The data is the following format:
input sequence name
POS VARV (H) FIRST_SEQ MOST_ABUND_SEQ CONSENSUS
_________________________________________________________________
1 0.191 L L L
2 0.000 L L L
3 0.323 E E E
4 2.121 N K c
5 0.323 S S S
6 0.191 Q Q Q
7 0.855 N N N
8 0.000 Q Q Q
9 0.000 Q Q Q
10 0.191 E E E
11 0.323 K K K
12 0.000 N N N
13 0.000 E E E
14 0.523 Q Q Q
15 0.191 E E E
|