BACKGROUND
USER GUIDE
BACKGROUND
Sequence variability within related protein sequences has
long ago been recognized to show significant clues about their 3-dimensional
structure and function. Sequence variability through mutation and subsequent immune
selection (a process called "antigen drift") is also a common mechanism by which
some viruses (AIDS, Influenza) and other pathogens (Plasmodium, Trypanosomatides, etc)
escape the immune system. Thus, for vaccine design, it is imperative to focus on protein fragments of
low variability (Shannon Entropy). Wu and Kabat (1977) were the first to define a
variability coefficient that led them to predict that
segments of high amino acid variability in immunoglobulins
correspond to their antigen binding
sites. Thus, high variability regions in a group of related proteins are linked to the
specificity of the molecules. On the other hand, regions with low variability are usually structurals,
or define regions of common function.
Shannon Entropy
Shannon entropy analysis (Shannon, 1948 ) is possibly the most sensitive
tool to estimate the diversity
of a system.
For a multiple protein sequence alignment
the Shannon entropy (H) for every position is as follow:
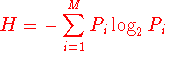
Where Pi is the fraction of residues of amino acid type
i, and M is the number of amino acid types (20).
H ranges from 0 (only one residue in present at that position) to 4.322
(all 20 residues are equally represented in that position). Typically,
positions with H >2.0 are considerered variable, whereas those with H < 2
are consider conserved. Highly conserved positions are those with H <1.0 (Litwin and Jores, 1992).
A minimum number of sequences is however required (~100) for H to describe the
diversity of a protein family.
USERGUIDE
Input
The input for this server must be a multiple sequence alignment in Clustal
format. Only the standard 20 amino acids should be included in the
alignment. If other sequence characters are included (e.g. X) the server
will return an error message.
A typical example of Clustal alignment is the following:
CLUSTAL W (1.81) multiple sequence alignment
hla_a68w_1HSB SHSMRYFYTSVSRPGRGEPRFIAVGYVDDTQFVRFDSDAASQRMEPRAPWI
hla_a0201_1DUY SHSMRYFFTSVSRPGRGEPRFIAVGYVDDTQFVRFDSDAASQRMEPRAPWI
hla_b3501_1A1N SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRTEPRPPWI
hla_b5301_1A1M SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRTEPRPPWI
hla_b5101_1E27 SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRTEPRAPWI
hla_b2701_1HSA SHSMRYFHTSVSRPGRGEPRFITVGYVDDTLFVRFDSDAASPREEPRAPWI
hla_cw3_1EFX SHSMRYFYTAVSRPGRGEPHFIAVGYVDDTQFVRFDSDAASPRGEPRAPWV
hla-cw4_1IM9 SHSMRYFSTSVSWPGRGEPRFIAVGYVDDTQFVRFDSDAASPRGEPREPWV
mkb_2vaa PHSLRYFVTAVSRPGLGEPRYMEVGYVDDTEFVRFDSDAENPRYEPRARWM
db-1BZ9 PHSMRYFETAVSRPGLEEPRYISVGYVDNKEFVRFDSDAENPRYEPRAPWM
.**:*** *::* ** **::: *****:. ******** . * *** *:
hla_a68w_1HSB RNTRNVKAQSQTDRVDLGTLRGYYNQSEAGSHTIQMMYGCDVGS
hla_a0201_1DUY GETRKVKAHSQTHRVDLGTLRGYYNQSEAGSHTVQRMYGCDVGS
hla_b3501_1A1N RNTQIFKTNTQTYRESLRNLRGYYNQSEAGSHIIQRMYGCDLGP
hla_b5301_1A1M RNTQIFKTNTQTYRENLRIALRYYNQSEAGSHIIQRMYGCDLGP
hla_b5101_1E27 RNTQIFKTNTQTYRENLRIALRYYNQSEAGSHTWQTMYGCDVGP
hla_b2701_1HSA RETQICKAKAQTDREDLRTLLRYYNQSEAGSHTLQNMYGCDVGP
hla_cw3_1EFX RETQKYKRQAQTDRVSLRNLRGYYNQSEAGSHIIQRMYGCDVGP
hla-cw4_1IM9 RETQKYKRQAQADRVNLRKLRGYYNQSEDGSHTLQRMFGCDLGP
mkb_2vaa RETQKAKGNEQSFRVDLRTLLGYYNQSKGGSHTIQVISGCEVGS
db-1BZ9 RETQKAKGQEQWFRVSLRNLLGYYNQSAGGSHTLQQMSGCDLGS
:*: * : * * .* ***** *** * : **::*.
Options
Plot variability.
Plot variability consists of a graph of the sequence variability plotted against the selected output sequence as shown below.
Mask variability in sequence
This option results in masking in the selected output sequence of those residues with a variability greater or equal than the selected variability threshold (see System and Methods). The variability masked sequence is returned in FASTA format (Shown below), and it can be used combination with the algorithm RANKPEP for the anticipation of conserved T-cell epitopes.
Conserved Fragments
This option identifies those fragments (minimum length selected by user) in the selected output sequence consisting of consecutives residues whose variability is under the set variability threshold. These fragments are returned sorted in a table by their position in the sequence alignment. Since sequence variability provides a means by which some pathogens escape the immune system, this option and that of the sequence variability masking are relevant for vaccine design considerations. It is important however to notice that relevant antigenic regions can be composed of conserved and variant regions. Unfortunatelly, these fragments will not appear in the conserved fragments ouput if they do not have the minimum number of consecutives conserved residues selected by the user.
Map structural variability
Mapping the sequence variability onto a representative 3D-structure requires providing the relevant PDB, and is carried out. At this moment, residue numbering in PDB must be consecutive and without missing or repeated residue numbers. Furthermore, the input alignment must be edited so that it is ungapped with regard to the sequence for which the 3D-coordinates are provided. In future releases, we plan to enhance SVA to save users from this manual-editing step. The output to this choice allows the visualization of the sequence variability onto the provided 3D-structure. Visualization requires prior installation of CHIME (www.mdl.com) in the browser as a helper plugging application. CHIME is a popular web based program for molecular graphics providing numerous options for visualization of 3D-structures. CHIME can also handle command scripting, and therefore we provide a window to allow users to enter their own scripts and/or commands. For those less versed in the use of CHIME, we provide a few action bottoms to display the variability onto several rendering of the 3D structure. Of course, CHIME permits the users to save the PDB on their desktops. If CHIME is not properly installed, the sequence variability mapped PDB will be download automatically onto the user's desktop. Once on the desktop, users can employ their preferred graphics program to visualize the variability. Within Rasmol, a popular and simple molecular graphics program, variability is visualized by simply selecting temperature in the Colours menu. In RASMOL and CHIME, sequence variability is visualize in a color scale that goes from blue for constant residues to red for highly variable residues as shown below.
Shannon, C. E. (1948) The mathematical theory of communication.
The Bell system Technical Journal, 27, 379-423 & 623-656.
Kabat, E. A., Wu, T. T., and Bilofsky, H. (1977) Unusual distribution of amino acids
in complementarity-determing (hypervariable) segments of heavy and light chains
of immunoglobulins and their possible roles in specificity of antibody
combining sites.J. Biol. Chem.252, 6609-6616.
Litwin, S. and Jores, R. (1992) In theoretical and experimental insights
into immunology, (Edited by Perelson A. S. and Weisbuch G.), Springer-Verlag,
Berlin
Last change: June 2005
